Szenarien des praktischen Umgangs
Es sind drei Szenarien denkbar, wie mit den Beschlüssen des G-BA über die Nutzenbewertung von Arzneimitteln nach Ablauf des Unterlagenschutzes umzugehen ist:
1.) Alle bereits gefassten G-BA Beschlüsse werden aufgehoben - eine neue Beschlussfassung ist nicht möglich;
2.) Alle bereits gefassten G-BA Beschlüsse bleiben gültig - eine neue Beschlussfassung ist nicht möglich;
3.) Alle bereits gefassten G-BA Beschlüsse bleiben gültig - eine neue Beschlussfassung ist weiter möglich.
In der Folge soll in essayistischer Form ausgeführt werden, welche konkreten Folgen mit diesen theoretischen Szenarien verbunden wären. Eine formelle juristische Bewertung liegt demnach außerhalb der Zielsetzung des vorliegenden Artikels.
Szenario 1 – G-BA-Beschlüsse werden aufgehoben
Denkbar wäre eine Rechtsauslegung, wonach der Ablauf des Unterlagenschutzes, mit welchem die tatbestandlichen Voraussetzungen für die Nutzenbewertung nach § 35a SGB V entfielen, nicht nur eine neue Beschlussfassung verunmöglichen, sondern gar zu einer Ungültigkeit bzw. Aufhebung bereits gefasster Beschlüsse des G-BA führen würde (so bspw. Fiekas, 2019).
Damit entfiele gleichzeitig die Grundlage für die Erstattungsbeträge nach § 130b SGB V. Es würde sich demnach eine Reihe von Fragen stellen:
- Sind Erstattungsbetragsverhandlungen (z. B. nach Kündigung) weiterhin möglich?
- Wenn ja: Wie wäre in den Erstattungsbetragsverhandlungen mit einer veränderten Evidenzlage umzugehen - in Ermangelung eines neuen G-BA Beschlusses (vgl. Szenario 2)?
- Wenn nein: Bliebe der bereits vereinbarte Erstattungsbetrag weiterhin gültig? Oder würde eine zweite Phase der unregulierten Preissetzung durch den pharmazeutischen Unternehmer resultieren?
Klar ist, dass es dem Gesetzgeber bei Entwurf des AMNOG fernlag, nach Ablauf des Unterlagenschutzes eine zweite Phase unregulierter Preissetzungen zu schaffen. Es bestehen daher anhand des Regelungsziels, aber auch anhand des Gesetzeswortlautes begründete Zweifel, ob dieses Szenario mit den derzeitig gültigen gesetzlichen Regelungen in Einklang zu bringen wäre.
Szenario 2 – G-BA Beschlüsse bleiben bestehen, können aber veralten
In einem zweiten Szenario wird davon ausgegangen, dass der Ablauf des Unterlagenschutzes zwar dazu führt, dass alle bereits gefassten Beschlüsse über die Nutzenbewertung ihre Gültigkeit beibehalten würden, allerdings keine neuen Beschlüsse gefasst werden könnten. Im Falle beispielsweise
- der Zulassung eines neuen Anwendungsgebiets,
- der Überschreitung der 50 Mio. €-Umsatzgrenze für Arzneimittel für seltene Leiden,
- des Ablaufs einer Befristung (z. B. aufgrund ausstehender Studienergebnisse) oder
- neuer wissenschaftlicher Erkenntnisse
könnte die bestehende Fassung der Arzneimittel-Richtlinie somit nicht aktualisiert werden und würde veralten bzw. wissenschaftlich obsolet werden.
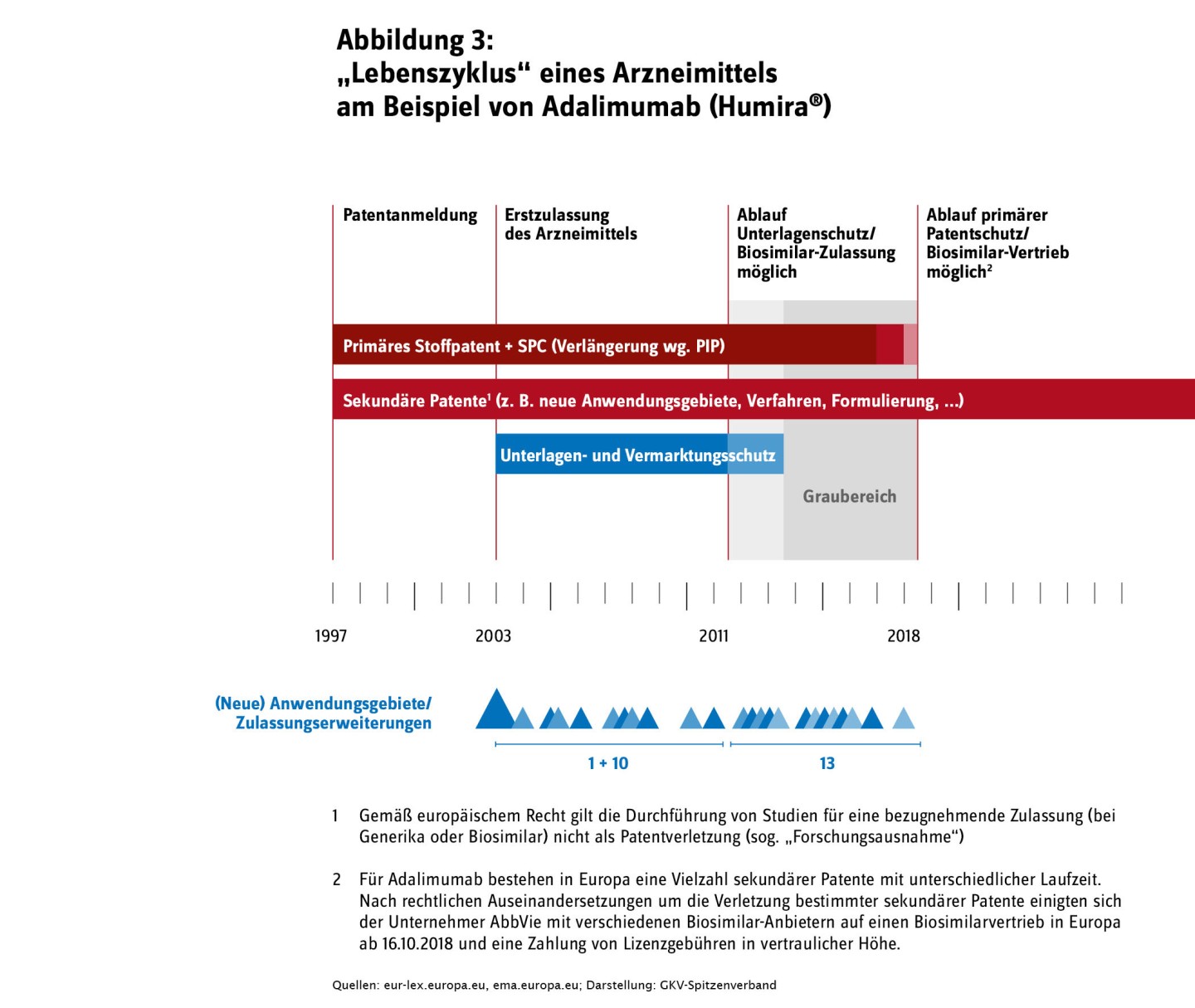
Wie das Beispiel Adalimumab eindrücklich zeigt, wird auch nach Ablauf des Unterlagenschutzes für ein dann noch patentgeschütztes Arzneimittel zum Teil erhebliche neue wissenschaftliche Evidenz generiert, die in der medizinischen Wissenschaft bislang nicht bekannt war. Es ist mittlerweile gefestigte und durch aktuelle Gesetzesnovellen (zuletzt beispielsweise der Einführung eines Arztinformationssystems mit dem GKV-Arzneimittelversorgungsstärkungsgesetz, AMVSG) regelmäßig bestätigte Rechtsaufassung, dass die Nutzenbewertung neben einer nutzenadäquaten Preisbildung für (patent-)geschützte Arzneimittel auch den Zweck hat, behandelnde Ärztinnen und Ärzte sowie Patientinnen und Patienten aktuell, transparent und neutral über den patientenrelevanten Nutzen dieser Arzneimittel zu informieren (vgl. Deutscher Bundestag, 2016). Vor diesem Hintergrund scheint auch Szenario 2 nur schwer mit den Zielen des Gesetzgebers in Einklang zu bringen.
Da die Erstattungsbeträge auf Basis des G-BA-Beschlusses zu verhandeln sind, würde dieses Szenario für die Verhandlungen der Erstattungsbeträge bedeuten, dass Kündigungen und Neuverhandlungen von bereits bestehenden Vereinbarungen auf Basis der gültigen Beschlüsse des G-BA weiterhin möglich wären.
Es ist allerdings derzeit vollkommen ungeklärt,
- wie in den Erstattungsbetragsverhandlungen mit wissenschaftlich obsoleten Beschlüssen umzugehen ist und in der Folge,
- ob bzw. wie sich Veränderungen der wissenschaftlichen Datenlage zu einem Arzneimittel, die nicht in einem neuen G-BA-Beschluss münden (können), preislich auf den Erstattungsbetrag auswirken können bzw. sollen.
Vorstellbar wäre, dass der auf Basis der bestehenden Beschlüsse verhandelte Erstattungsbetrag auch für alle neu zugelassenen Anwendungsgebiete des Arzneimittels gelten würde und Veränderungen der wissenschaftlichen Datenlage zu bestehenden Anwendungsgebieten keine Berücksichtigung bei erneuten Erstattungsbetragsverhandlungen fänden. Eine solche einfache Übertragung würde dazu führen, dass der Erstattungsbetrag in manchen Fällen niedriger läge als angemessen, in anderen Fällen wiederum höher. Beispielsweise zeigte sich in der Vergangenheit, dass ein Zusatznutzen zum Teil erst anhand von langfristigen Studienergebnissen nachgewiesen werden kann (z. B. bei Ergebnissen im Gesamtüberleben bei niedrig-malignen Tumorerkrankungen nach einer Zulassung auf Basis von Surrogatendpunkten). Dürften diese positiven Langzeit-Ergebnisse keine Berücksichtigung finden, weil ein neuer G-BA Beschluss aufgrund des zeitweiligen Ablaufes des Unterlagenschutzes ausgeschlossen wäre, so würde für das Arzneimittel ein unangemessen niedriger Erstattungsbetrag gelten.
Theoretisch wäre diese Lösung zudem strategieanfällig: Ist beispielsweise für ein Arzneimittel um den Zeitpunkt des Ablaufs des Unterlagenschutzes herum die Zulassung neuer Anwendungsgebiete geplant, so ergäbe sich der Anreiz, Anwendungsgebiete, für die ein Zusatznutzen oder eine hohe Teilpreiskomponente absehbar sind, eher vor Ablauf des Unterlagenschutzes, solche für die kein Zusatznutzen oder ein niedriger Teilerstattungsbetrag droht, eher nach Ablauf des Unterlagenschutzes zur Zulassung zu bringen.
Eine weitere angenommene Konstellation läge darin, dass in Ermangelung einer erneuten G-BA-Nutzenbewertungen eine separate, „kleine Nutzenbewertung“ durch die Industrie oder den GKV-Spitzenverband initiiert würde, indem die wissenschaftliche Bewertung der veränderten Datenlage in die Erstattungsbetragsverhandlungen bzw. das Schiedsverfahren verlagert würde. Eine solche Lösung würde eklatant dem Grundprinzip des AMNOG-Verfahrens, nämlich der klaren Trennung des evidenzbasierten Bewertungsverfahrens eines Arzneimittels einerseits und der anschließenden Preisverhandlungen andererseits, widersprechen. Für die Qualität, Glaubwürdigkeit, Transparenz und Rechtssicherheit des Bewertungsverfahrens maßgebliche Verfahrensschritte wie die vollständige Aufarbeitung der zugrundeliegenden Evidenz, die gutachterliche Bewertung durch ein unabhängiges Institut sowie das schriftliche und mündliche Stellungnahmeverfahren entfielen in einem solchen Szenario gänzlich.
Ebenso erscheint zumindest möglich, dass für nach Ablauf des Unterlagenschutzes neu zugelassene Anwendungsgebiete der ursprüngliche frei gewählte Abgabepreis des pharmazeutischen Unternehmers oder ein anderer durch den pharmazeutischen Unternehmer zu bestimmender unregulierter Preis gelten würde. Dieser Preis müsste dann mit den bestehenden
(Teil-)Erstattungsbeträgen zu den bereits bewerteten Anwendungsgebieten zu einem Mischpreis kalkuliert werden (GKV-Spitzenverband, 2018). Es ist offensichtlich, dass durch einen willkürlichen Teilpreis die Mischpreissystematik ad absurdum geführt würde: Durch einen ausreichend hohen Teilpreis für das neue Anwendungsgebiet ließe sich jeder gewünschte Mischpreis erreichen. Auch dieses Szenario liefe demnach auf eine zweite Phase der unregulierten Preissetzung hinaus und widerspräche demnach fundamental der Intention des Gesetzgebers.
Szenario 3 – Komplementarität aus AMNOG-Verfahren und Festbetrag
Wie weiter oben ausgeführt wurde das AMNOG-Verfahren als Ergänzung zur Festbetragsregelung entworfen, um bestehende Regelungslücken zu schließen. Es liegt demnach nahe, dieser Komplementarität auch in den tatbestandlichen Voraussetzungen Rechnung zu tragen und damit Situationen, in denen ein Arzneimittel weder dem einen noch dem anderen Regulativ unterliegt, effektiv zu verhindern. Andernfalls wäre zu befürchten, dass die oben beschriebenen Regelungslücken zulasten der Beitragszahlenden genutzt würden.
In Szenario 3 soll demnach davon ausgegangen werden, dass ein Arzneimittel solange dem AMNOG unterliegt, wie für dieses Arzneimittel kein Festbetrag gilt. Bestehende Beschlüsse zur Nutzenbewertung würden fortgelten und neue Beschlüsse könnten (bspw. im Falle einer neuen wissenschaftlichen Datenlage) unverändert gefasst werden. Damit ließe sich einerseits sicherstellen, dass den medizinischen Fachkreisen sowie Patientinnen und Patienten für nicht festbetragsgeregelte Arzneimittel deren transparent und neutral bewertete Datenlage ständig aktuell zur Verfügung stünde. Auch der Erstattungsbetrag wäre damit dem aktuellen Wissen zum Nutzen dieses Wirkstoffes angemessen. Arzneimittel, die auch nach Ablauf des Unterlagenschutzes nicht in die Festbetragsregelung einbezogen werden können (bspw. weil wegen eines bestehenden Patentschutzes noch keine Generika oder Biosimilars auf dem Markt sind oder weil bei Einbeziehung in eine Festbetragsgruppe erforderliche Therapiemöglichkeiten eingeschränkt würden), würden damit weiterhin dem Preisregulativ der Erstattungsbeträge unterliegen. Dieses Vorgehen ist notwendig, da solche Arzneimittel weiterhin eine marktexklusive Stellung besitzen. Somit ließe sich durch die Kombination der Instrumente Festbetrag und Erstattungsbetrag eine annähernd lückenlose Regulierung des Arzneimittelmarktes sicherstellen.
Um diese Lösung allerdings Realität werden zu lassen, bedarf es einer Klarstellung des Begriffes des neuen Wirkstoffes in der Arzneimittel-Nutzenbewertungsverordnung (AM-NutzenV) durch den Gesetzgeber. Demnach muss ein Wirkstoff solange als neuer Wirkstoff gelten, wie für ihn noch kein Festbetrag gilt.