Arzneimittel
Die Neuordnung der Hämophilie-Versorgung mit dem Gesetz für mehr Sicherheit in der Arzneimittel-Versorgung
Im Februar 2018 erhielt der Wirkstoff Emicizumab seine Zulassung zur Therapie bei Hämophilie A und wurde im deutschen Markt unter dem Handelsnamen Hemlibra in Verkehr gebracht. Dieser Markteintritt sollte ein Gesetzgebungsverfahren anstoßen, das den regulativen Rahmen für die Versorgung in der Hämophilie in Deutschland fundamental reformieren würde.
Inhalt
- Das Krankheitsbild der Hämophilie
- Von der Krankenhausbehandlung in die ärztlich kontrollierte Heimselbstbehandlung
- Der „Stein des Anstoßes“ für einen anderen Vertriebsweg
- Mit dem GSAV neue Wege in der Hämophilie-Versorgung beschreiten
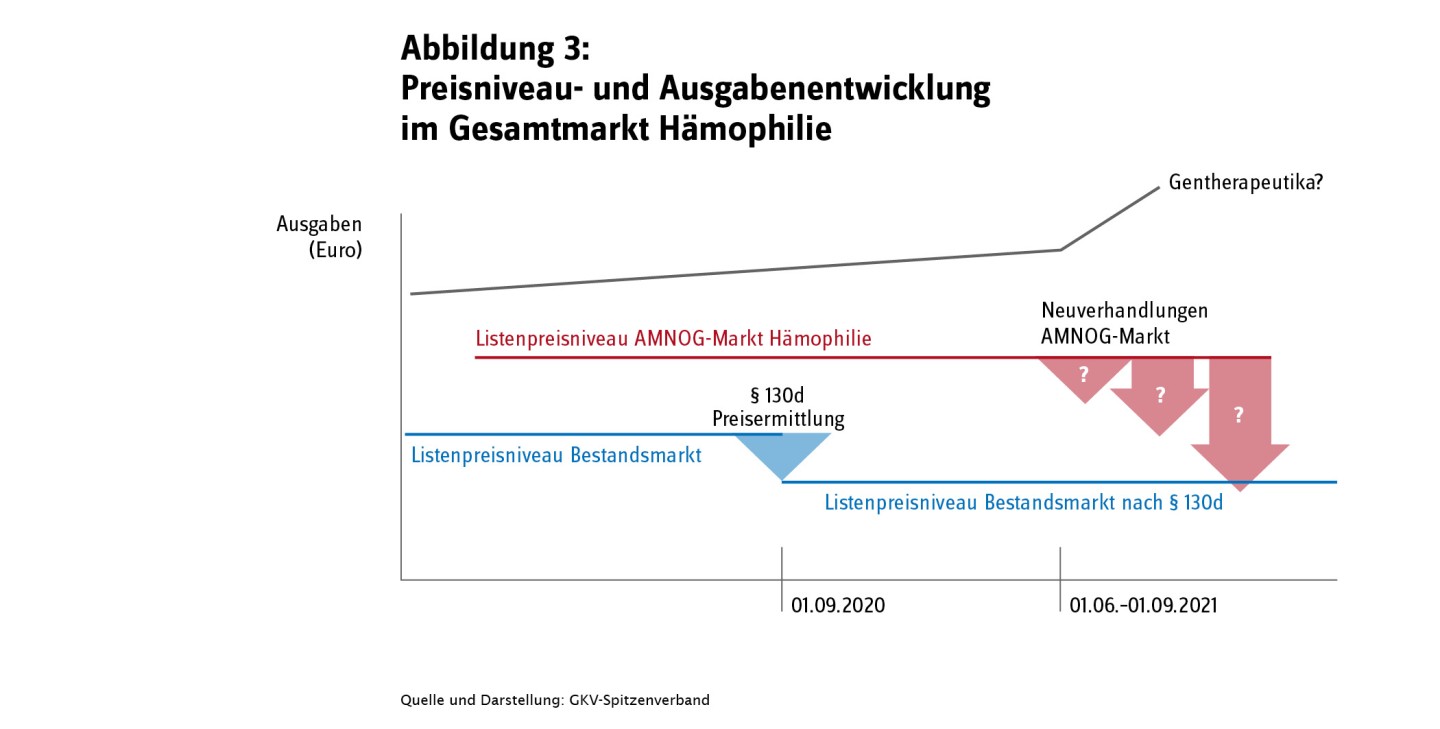
- Die Preisneuordnung im Hämophilie-Markt
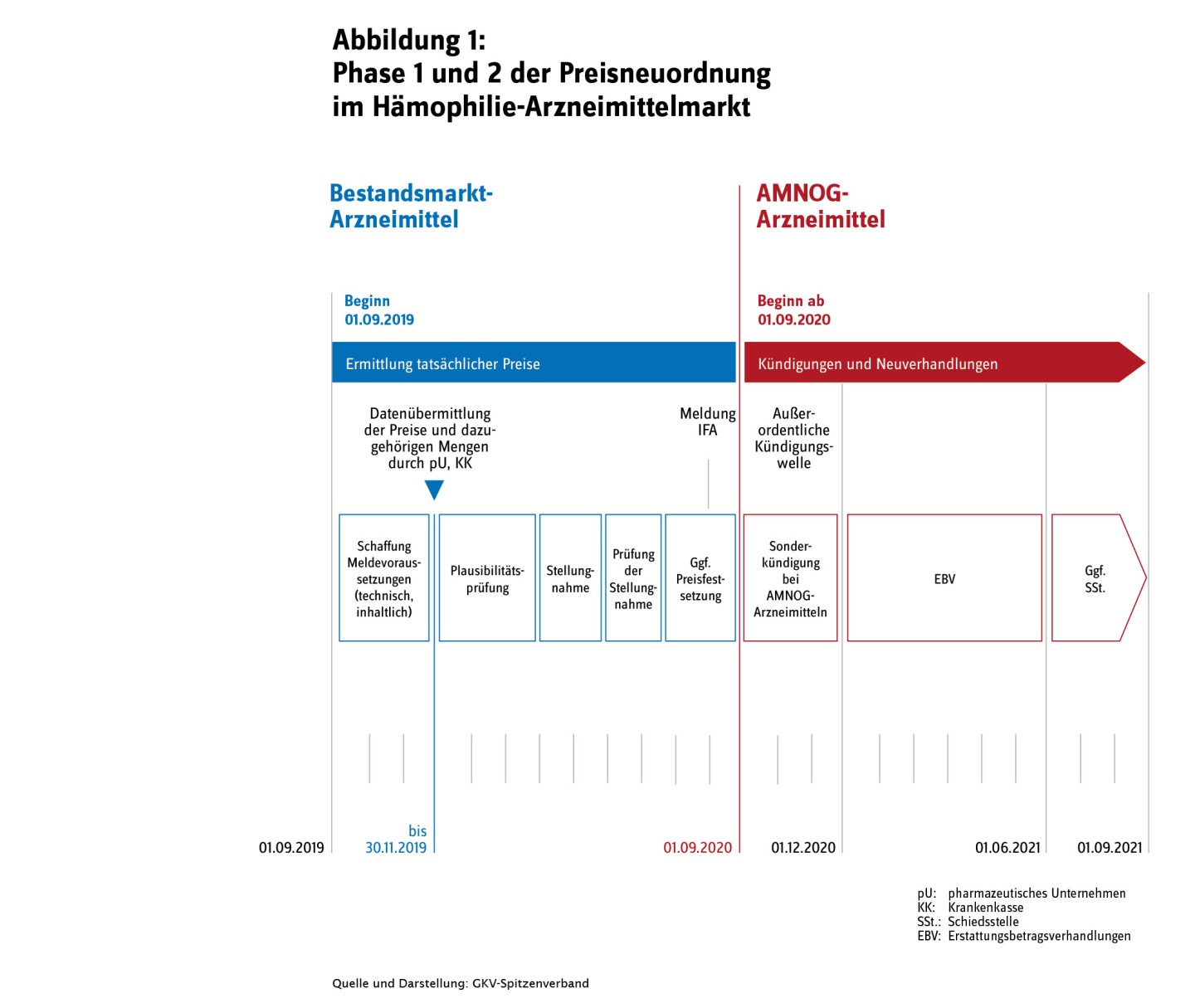
- Phase 1: Preisneuordnung führt Bestandsmarkt in die Transparenz
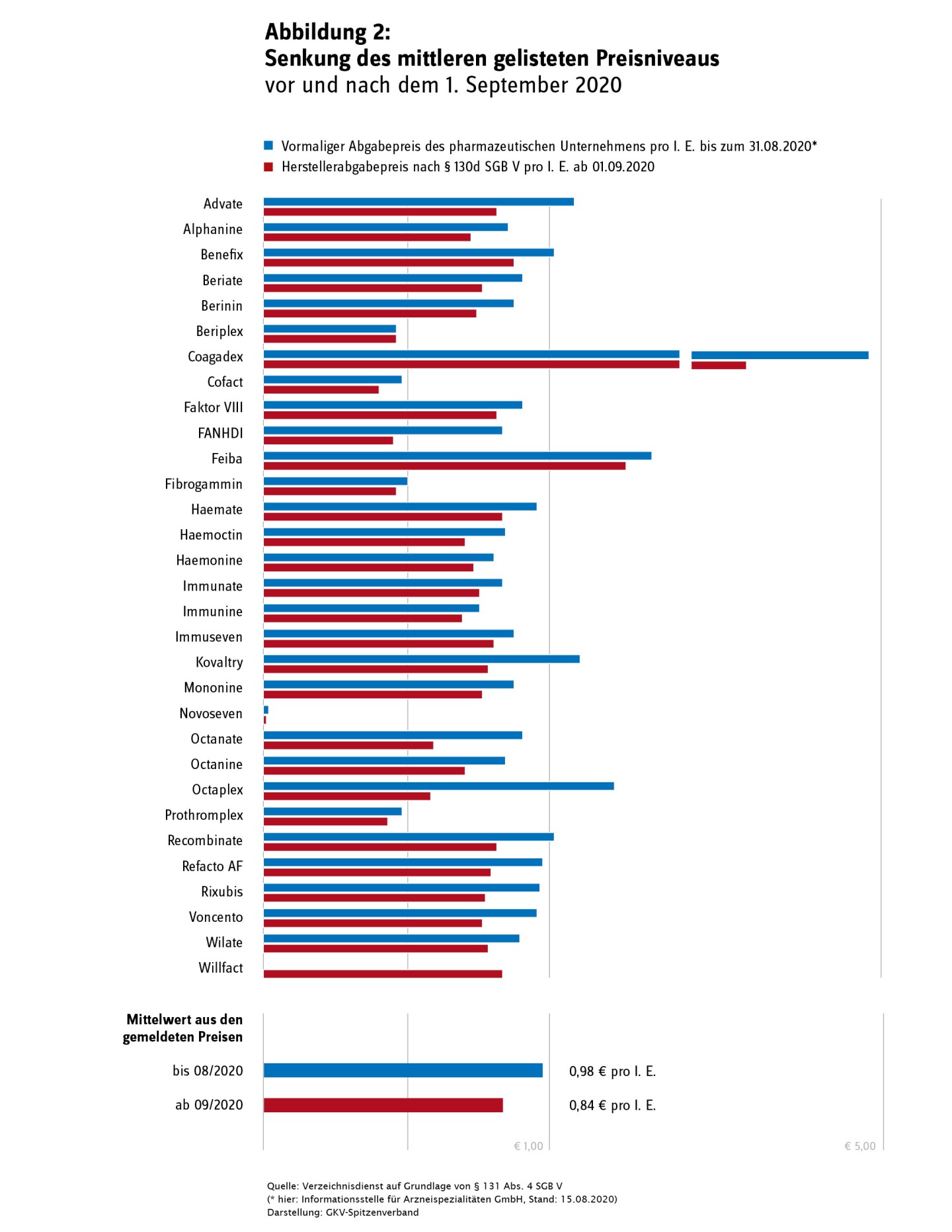
- Ergebnisse der Preisneuordnung im Bestandsmarkt
- Phase 2 der Preisneuordnung des Hämophilie-Marktes läuft seit 1. September 2020
- Vorteile der Neuordnung für Betroffene
- Sicherung und Fokussierung bewährter Versorgungsstrukturen in der Hämophilie auf die Behandlungsqualität
- Unmittelbarer Zugang der GKV zum vollen Datensatz des DHR
- Weiterentwicklung des Deutschen Hämophilieregisters zu einem AbD-tauglichen Register
- Fazit und Ausblick: Der weitere Weg
- Literaturverzeichnis